产品规格:
产品数量:
包装说明:
关 键 词:芜湖MDR2017/745厂
行 业:咨询 管理咨询
发布时间:2022-12-14
评估及更新质量管理体系(以下第3点) 检查可用证据和风险管理的充分性,识别差异(第56条) 评估产品标签(附件I第Il1章) 确保上市后监督的安排充分适宜够(第七章第1节) 制定上市后性能跟踪(PMCF,附件XIV B部分) 做好迎接新的警戒需求的规定(第七章第二节) 确保可追溯相关方面的义务(第3章) 3,质量体系评估 评估新IVDR法规下QMS符合标准和流程的程度 增加新法规应用于QMS的要求 协助和识别合规负责人(PPRC)并参与培训 4,公告机构NB确认 选择合适的公告机构,确认公告机构资质及范围
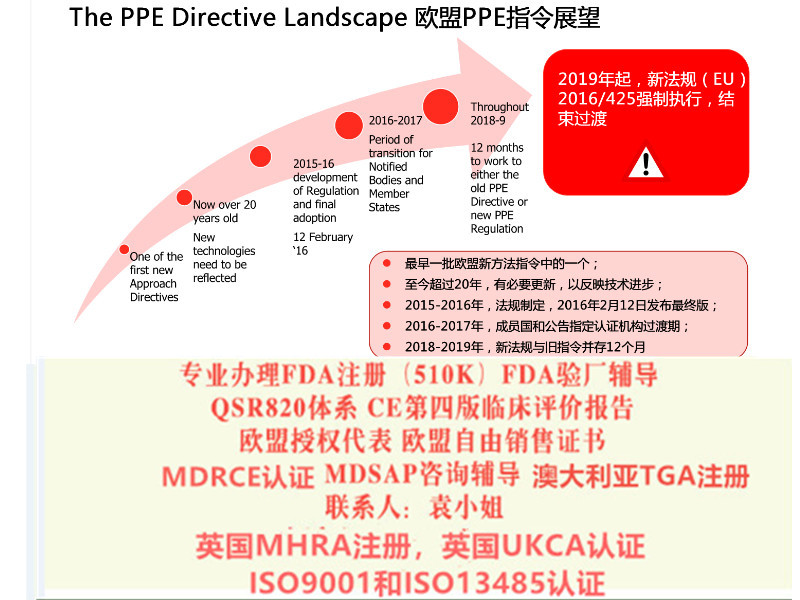
新的器械法规(2017/745 / EU)(MDR)和体外诊断器械法规(2017/746 / EU)(IVDR)使欧盟立法与技术进步,科学变革和进步在法律制定上保持一致。
新法规将建立一个健全,透明,可持续的框架,得到国际认可,可提高安全性并为制造商创造公平的市场准入。与指令不同,法规不需要转变为国家法。
器械法规(MDR)的背景
MDR将取代现有的器械指令(93/42/EEC)(MDD)和主动植入式器械指令(90/385/EEC) (AIMDD)。MDR于2017年5月发布,标志着MDD和AIMDD之间为期三年的过渡期的开始。
在过渡期间,MDR将逐步生效,首先是与公告机构和制造商根据MDR申请新证书的能力有关的规定。
上市后跟踪的规定,要求制造商主动收集和评估上市后数据,旨在确认器械的有效性、识别之前未知的并已识别的和禁忌症、识别并分析突发风险、确保收益/ 风险的可接受性以及确定器械可能的操作不当或超标示使用以验证其预期用途是否正确。上市后跟踪在产品生命周期中的作用不可忽视。
关于MDR 涵盖产品范围和分类规则法规对“器械”的定义结合了90/385/EEC 和93/42/EEC 指令的范围,并有所扩大,纳入了有源植入器械、软件以及植入或侵入人体的非用途产品,如眼镜、美容植入物、去除脂肪组织的器械、发射高强度电磁进行皮肤等操作的器械、利用磁场大脑元活动的器械等。对药械组合产品的描述也更为具体,法规还适用于使用活性或非活性动物、人源组织或细胞的产品制造而成的器械。器械的分类仍延续了之前的大类,即按照风险等级分为类:Ⅰ、Ⅱ a、Ⅱ b、Ⅲ类。但分类规则较之前有所增加,由18 条增加至22 条。具体分类规则条款情况及变化情况见下表2:欧盟器械新法规MDR主要变化情况介绍
在此基础上,会议对于MDR及IVDR的执行达成了如下时间表。2016年12月14日,EUDAMED(European databank for medical devices) 筹划会上欧盟各国对于器械法规MDR及IVDR的执行进行了一轮的讨论,与会人员对于这两个法规的细节内容进行了讨论并达成了一致意见。在此基础上,会议对于MDR及IVDR的执行达成了如下时间表。一,DMR的主要变化1.扩大了应用范围2.提出了新的概念和器械的定义3.细化了器械的分类4.完善了器械的通用和性能要求5.加强对技术文件的要求6.加强器械上市后的7.完善评价相关要求8.提出Eudamed数据库的建立和使用9.提出器械的可追溯性(UDI)10.对NB提出严格的要求MDR简介2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。MDR将取代Directives 90/385/EEC (有源植入类器械指令)and 93/42/EEC(器械指令)。
英国已经脱欧了,不再认可欧洲的CE证书,需要UKCA认证,有英代,MHRA注册,才可以合规出口英国。