产品规格:
产品数量:
包装说明:
关 键 词:郴州2017/745厂
行 业:咨询 管理咨询
发布时间:2022-12-06
但是新MDR的管控趋于严格,对于制造商,欧盟授权代表以及国外进口商三方该承担的责任比较明确,欧盟授权代表和进口商与制造商一样为缺陷器械承担连带的法律责任。所以进口商在采购工厂产品的时候,较MDD老法规,他们更关注,工厂是否真正满足CE法规要求,尽量的将自己要承担的风险降低到。
上市后跟踪的规定,要求制造商主动收集和评估上市后数据,旨在确认器械的有效性、识别之前未知的并已识别的和禁忌症、识别并分析突发风险、确保收益/ 风险的可接受性以及确定器械可能的操作不当或超标示使用以验证其预期用途是否正确。上市后跟踪在产品生命周期中的作用不可忽视。
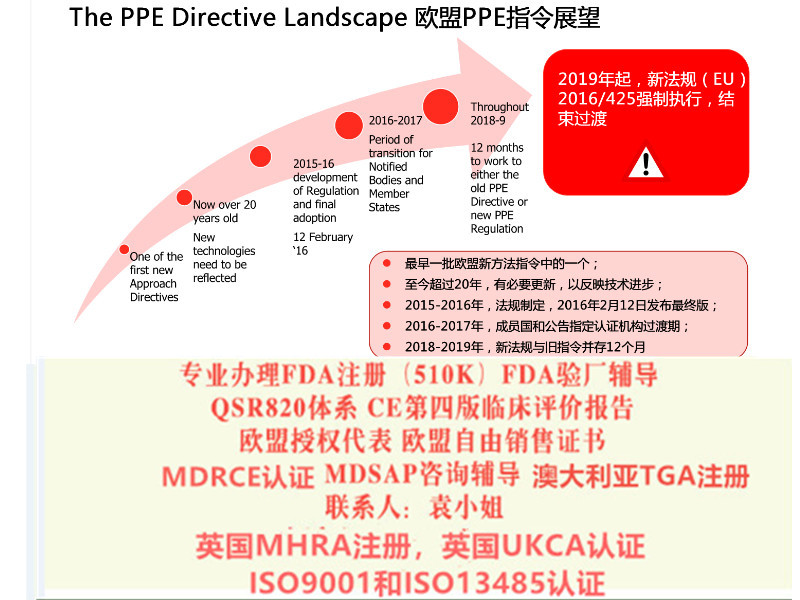
2017年5月5日,欧盟正式发布了OfficialJournal其正式对外宣布了新版MDR(REGULATIONEU2017/745)法规和新的IVDR(REGULATIONEU2017/746)法规。新法规将取代现行的三个器械指令:分别是器械指令93/42/EEC,有源器械指令90/385/EEC及体外诊断器械指令98/79/EEC。
总的说来新的MDR和IVDR加强了体系管理,对高风险设备增加了相关规定比如对于非用途但具有与器械相似特性的设备也将受到新法规的管辖(如用于美容的彩色隐形眼镜),提升了产品对患者的透明度和可追溯性并设立电子数据库(Eudamed)、设备将有一个的识别号这加强其在整个供应链的可追溯性。
SUNGO提醒我们的客户在申请产品CE认证时,在过渡阶段请谨慎考虑是选用法规还是采用老的指令方案,同时也需要对NB机构的发证进行了解和确认以保证产品在欧盟市场销售的可延续性。
我司注意到新法规主要在以下几点上发生了变化:
1.器械的定义;
2.器械的分类;
3.基本安全和性能要求;
4.技术文件要求;
5.评价;
6.上市后;
7.Eudamed数据库;
8.对NB公告机构的要求(新法规生效后NB将按照新的要求重新进行授权);
9.对高风险器械的新增了要求;
公告机构可于2017年11月26日起申请。的过程可能需要12个月或更长时间,包括来自不同和欧洲当局的评估人员。这意味着,根据新规定的批公告机构可能在2018年底前到位。公告机构数据库(NANDO)可在此找到。
ISO13485认证,CE评估报告编写,FDAQSR820等产品出口的相关认证