瑞士法规价格 瑞士代表CHRN批发 如何办理瑞代
价格:10000.00起
产品规格:
产品数量:
包装说明:
关 键 词:如何办理瑞代
行 业:咨询 管理咨询
发布时间:2022-11-04
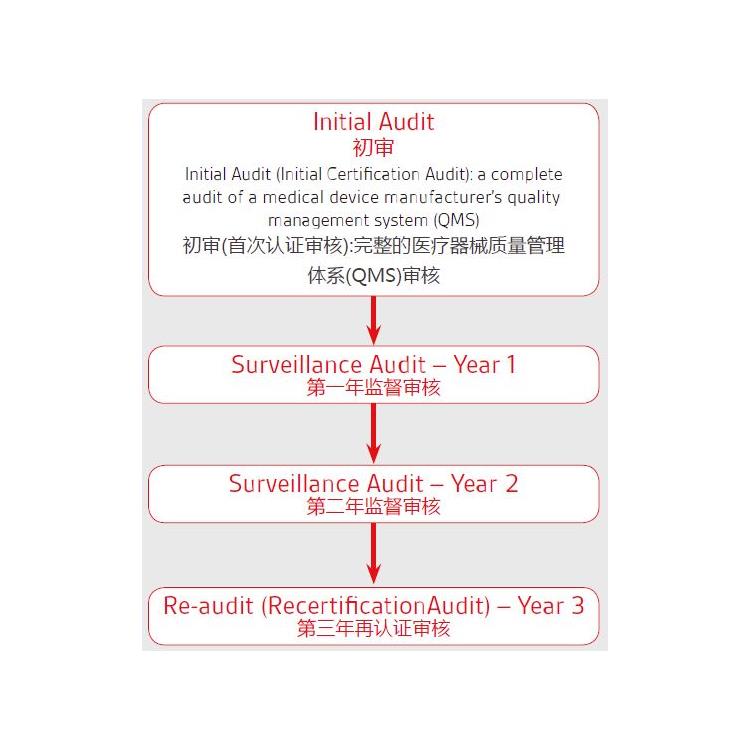
瑞士技术文件编写的流程和周期
1) 收集编写技术文件需要提供的资料:企业信息、产品说明书、产品测试报告、产品包装标签图片、产品上市后信息等;
2) 进行文件的编写;
3) 文件编写好进行内部评审(I类)或交由第三方认证机构评审(高风险器械);
4) 评审提出的不符合项整改;
5) 完成整改继续评审没问题后交付企业。
周期:I类器械4-8周,高风险器械12-24周(具体时间由企业配合提供资料的进度决定)。
中国制造商出口器械产品到瑞士,需要满足什么条件?
A1:瑞士对于器械的管理规定和欧盟基本相同。依据器械分类不同采取自我宣告和合格 评定两种模式。其中合格评定认可欧盟公告机构的证书,自我宣告也需要企业编制技术文件 和 DOC。除此之外,制造商需要瑞士代表,但是不需要进行器械注册。总结如下: 1) 编制技术文件和 DOC;
2) 提供 MDR/IVDR 的 CE 证书(非自我宣告产品适用);
3) 瑞士代表,并将 1)和 2)文件提交瑞代;
4) 标签/说明书/外包装/器械伴随文件等加贴瑞代信息;
5) 产品出口以及上市后管理。
协助判定产品分类
1) I类器械:通常是指不接触人体或只接触完整皮肤的器械。
2) I-m(测量)器械:带有测量功能的I类器械。
3) I-s()器械:终以形式出现在市场上的I类器械。
4) IIa类器械:风险等级较一类器械高,一般是指暂时使用的侵入器械等,有能量交换或测量的有源器械。
5) IIb类器械:风险等级较高,一般指会对人体有潜在风险或者是长时间使用。
6) III类器械:风险等级,一般用于人体循环系统或大脑。
补充:瑞士立后,目前的法规在器械上更趋近于欧盟的MDR法规,而在体外诊断器械上的要求更趋近于IVDD法规的要求。
瑞士授权代表的流程和周期
1) 提供企业基本信息和产品信息;
2) 双方签订瑞士授权代表协议;
3) 委托方将文件放在瑞士技术文档里。
周期:瑞士授权代表协议签订周期一般较快,受托方搜集了委托方的技术文件和基本信息后即可签订协议,协议经双方签字盖章后即可生效。
瑞士授权代表办理的时机
所有器械在进入瑞士市场的前提是完成瑞士注册。
但是,非瑞士制造商将需要一名瑞士授权代表来完成注册。
未能注册器械意味着不能合法地出口。
具体的截止日期可同时参考“瑞士技术文件编写的时机”中a)段的表格。
SUNGO致力于为全球的器械生产商和经营者提供市场准入的合规咨询以及国际注册服务。从产品生产、检测、过程管理、注册、认证、整改、上市跟踪等各环节为企业提供的技术支持,为产品合规和顺利上市保驾护航。十多年里,SUNGO已为全球30多家上市公司和全球器械制造商,合计5000多家企业提供过相关服务。