产品规格:
产品数量:
包装说明:
关 键 词:出口瑞士需要办理MHRA如何办理
行 业:商务服务 认证服务
发布时间:2022-10-26
上市流程
1. 瑞士授权代表(仅适用于非瑞士经济运营体);
2. 在瑞士局进行经济运营商登记注册;3. 在瑞士局进行器械注册;4. 准备符合性声明/证书和技术文件;5. 更换产品包装;6. 加贴符合性标记;
PART2 什么时候需要瑞士代表?
2.1 制造商在欧盟/欧洲经济区内或者了欧盟授权代表
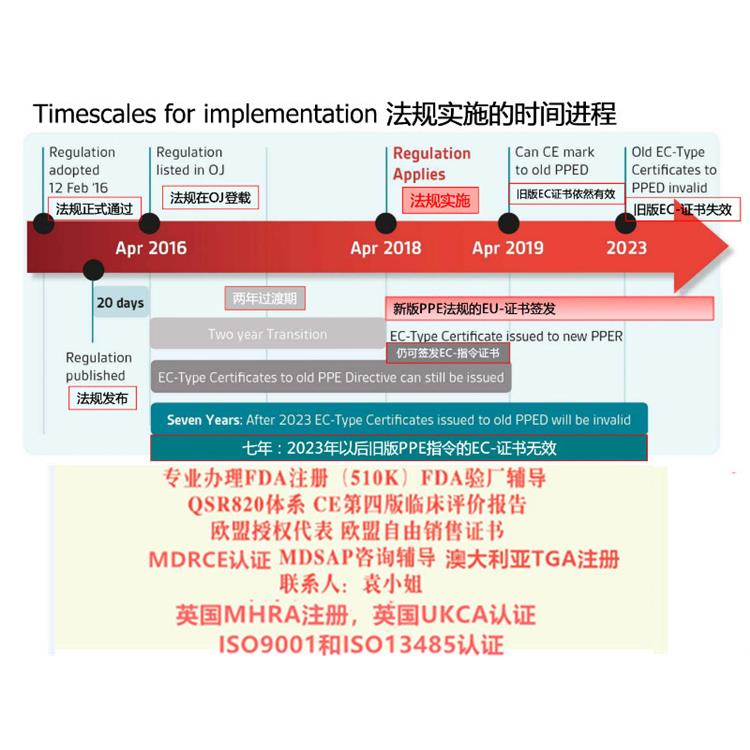
此时制造商享有过渡期,在过渡期截止日期之前瑞士代表即可。
对于III类器械、IIb类植入式器械和有源植入式器械:2021年12月31日
对于非植入式IIb类设备和IIa类设备:2022年3月31日
对于I类设备:截至2022年7月31日
对于系统和程序包必须在2022年7月31日前一名符合第51条第5款的授权代表。
2.2 如果制造商不在欧盟或欧洲经济区内,同时又没有在2021年5月26日之前欧盟授权代表的制造商,应在2021年5月26日或者进行瑞士器械贸易时瑞士代表。
在2021年5月26日之前,非欧盟向瑞士出口器械时,需要欧盟授权代表可以满足要求。
但是从2021年5月26日之后,出口瑞士需要单瑞士代表(CH-REP)。
PART1什么是瑞士代表?
瑞士代表的定义:任何在瑞士境内成立的自然人或法人已收到并接受位于瑞士的制造商的书面授权,以代表制造商其在器械条例下的义务的特定任务采取行动。
为了能够履行必要的职责,瑞士代表应其法规负责人PRRC。同时瑞士代表应确保具备充分的知识以应对器械上市前和上市后的各项工作
另外,按照瑞士法规要求,瑞士代表也需要在对瑞士的器械贸易发生后的三个月内完成其CHRN的注册。
欧盟法规已经升级了,您的产品属于I类吗?欧盟新法规要求非常严苛,您是否按照新法规MDR/IVDR办理了CE了?是否有欧盟代表,欧盟注册,SRN号码,Basic UDI,是否已经申报数据库Eudamed?
2:英国已经脱欧了,不再认可欧洲的CE证书,需要UKCA认证,有英代,MHRA注册,才可以合规出口英国。
3:瑞士也已经不认可欧洲的CE认证,您有产品出口瑞士吗?是否有做瑞士代表以及瑞士注册的?
4:手术衣510K,隔离衣510K,手套510K,电动/手动轮椅510K,FDA注册,FDA美国代理人
5:ISO13485认证,CE评估报告编写,FDAQSR820等产品出口的相关认证
协助判定产品分类
1) I类器械:通常是指不接触人体或只接触完整皮肤的器械。
2) I-m(测量)器械:带有测量功能的I类器械。
3) I-s()器械:终以形式出现在市场上的I类器械。
4) IIa类器械:风险等级较一类器械高,一般是指暂时使用的侵入器械等,有能量交换或测量的有源器械。
5) IIb类器械:风险等级较高,一般指会对人体有潜在风险或者是长时间使用。
6) III类器械:风险等级,一般用于人体循环系统或大脑。
补充:瑞士立后,目前的法规在器械上更趋近于欧盟的MDR法规,而在体外诊断器械上的要求更趋近于IVDD法规的要求。。
瑞士技术文件编写的时机
对于生产器械的企业,出口瑞士前技术文件编写的时机应分成2种情况来分别参考:
a) 如果制造商在欧盟/欧洲经济区内或者已欧盟授权代表,完成产品注册以及CE技术文件,此时制造商应在以下截止日期之前需要编写好技术文件:
产品/风险等级 截止日期
III类器械、IIb类植入式器械和有源植入式器械 2021年12月31日
非植入式IIb类设备和IIa类设备 2022年3月31日
I类设备 2022年7月31日