手套的CEMDR2017/745认证厂商
价格:面议
准备CE标记的技术文件(Technical Documentation)或设计文档(Design Dossier)。
办理欧盟CE认证(CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书
关于经济运营商各方义务
法规在第I章第2条定义中提出了“经济运营商”的概念,经济运营商是指制造商、授权代表、进口商、经销商以及任何对系统或手术包类器械进行组合或并投放市场的自然人或法人。即在符合法规规定情况下负责器械生产(包括组合或)、销售及上市后运营的自然人或法人。
法规先规定了制造商的义务,涵盖生产、合规、上市后的产品全生命周期,但法规同时规定,经销商、进口商或其他自然人或法人在向市场提供以其名字、注册商标命名的器械时应承担制造商相应的义务,也包括变更相应器械预期用途或变更其他影响其符合性的事项的情况。在上市后要求中,经济运营商同时负有相应的责任和义务。
法规对各方义务的描述更为明确也更为具体,对于制造商的要求更为细化,因此新法规执行后,各方应先明确自身职责和义务,规范有序地开展生产和市场活动,应审核确认上游供应商是否符合规定,并确认能够自己的下游流程符合规定,应按照对应的警戒系统的要求进行或配合事件上报,配合完成现场纠正措施,并依据职责组织培训。
四、其他
法规中规定了对于一次性使用器械的再处理即复用的要求。
MDR
办理欧盟CE认证(CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书
CE技术文件
CE技术文件是欧盟器械指令中很重要的一个事项,它的目的是要求企业准备充份的技术资料和,供主管机关抽查,或发生诉讼纠纷时使用。各欧盟指令对于"技术档案"的要求有所差别,在这里谨以中国出口企业常用的“器械”的要求为例,加以说明。
器械指令93/42/EEC要求"技术档案"可能包含:企业的质量手册和程序文件;企业简介及欧洲代表名称、联系方式;CE符合性声明(或称自我保证声明,若该产品是和其它设备联合运用,则应有整体符合基本要求的材料),主要内容如下:
1、产品名称、分类
2、产品概述(包括类型和预期用途)
◇产品的历史沿革
◇技术性能参数
◇产品配合使用的附件、配合件和其它设备清单
◇产品的图示与样品
◇产品所用原材料及供应商
3、使用该产品的调和标准/或其它标准
4、风险分析评估结论和预防措施
5、生产质量控制
◇产品资料和控制文档(包括产品生产工艺流程图)
◇产品的方法和确认的描述
◇验证
◇产品质量控制措施
◇产品稳定性和效期的描述
6、包装和标识
◇包装材料说明
◇标签
◇使用说明书
7、技术评价
◇产品检验报告及相关文献
◇技术概要及观点
8、风险管理
◇产品潜在风险报告及相关文献
◇潜在风险的概要及观点
9、评价
◇产品测试报告及相关文献
◇使用概述及观点
附1、产品出厂检测报告
附2、产品型式检测报告
附3、基本要求检查表
备注:
◇研究(包括:物理性能,生化、药理 、药动及毒性研究,测试,合格,相容性等)
◇生物兼容性测试(A)部分要求:细胞毒性、感光性、-皮内反应、急性全身中毒、致热性、亚急性中毒、遗传毒性、植入溶血性; (B)支持测试:慢性中毒、致癌性、再生性/生长性、生物动因退化。)
◇资料(需要研究或描述研究)
◇包装合格
◇标签、使用说明
◇结论(设计档案资料的接受、利益对应风险的陈述)
10、欧盟授权代表信息及协议
11、符合基本要求表
12、协调标准
13、警戒系统程序
2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。
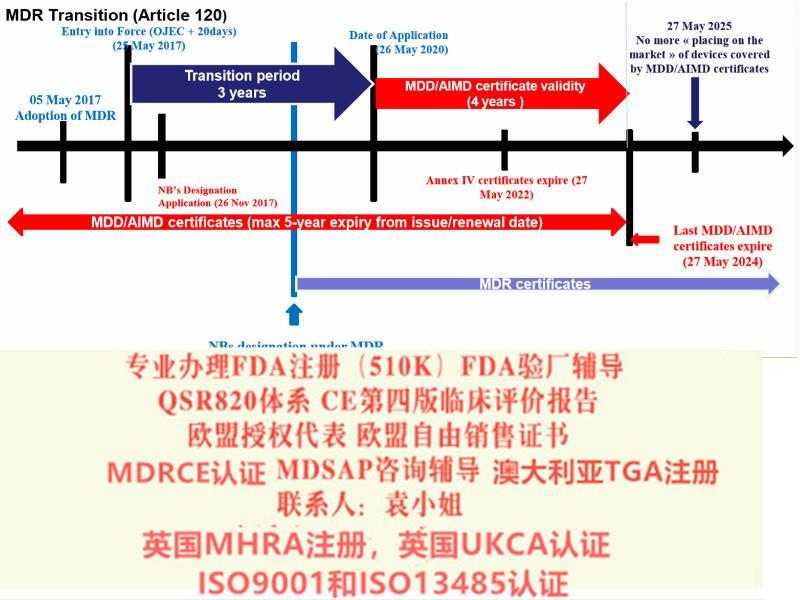
并可在2020 年5 月26 日前并通知符合新法规的符合性评估机构。公告机构可在2020 年5 月26 日前, 采用合规的符合性评估流程并按照新法规签发证书。对于特定Ⅲ类器械和Ⅱ b 类器械产品,在已委派必要的器械协调小组(MDCG)、小组前提下,同样可通过指令豁免在2020 年5 月26 日之前投放市场。法规关于公告机构的要求(正文第35~50 条) 自2017 年11 月26日起适用,即公告机构在新法规发布后的六个月内即应开始进行相应的申请,符合要求后方可依据新法规开展符合性评估。同时法规对成员国主管机构的和MDCG 的成立也设定了期限,要求于2017 年11 月26 日前完成。对于成员国主管机构之间的协调,设定期限为2018 年5 月26 日。
SUNGO服务的宗旨不仅是完成项目,而是帮助客户解决问题并达成目标。