产品规格:
产品数量:
包装说明:
关 键 词:助行器的SUNGO的欧代
行 业:咨询 管理咨询
发布时间:2022-08-27
1:欧盟法规已经升级了,您的产品属于I类吗?欧盟新法规要求非常严苛,您是否按照新法规MDR/IVDR办理了CE了?是否有欧盟代表,欧盟注册,SRN号码,Basic UDI,是否已经申报数据库Eudamed?
2:英国已经脱欧了,不再认可欧洲的CE证书,需要UKCA认证,有英代,MHRA注册,才可以合规出口英国。
3:瑞士也已经不认可欧洲的CE认证,您有产品出口瑞士吗?是否有做瑞士代表以及瑞士注册的?
4:手术衣510K,隔离衣510K,手套510K,电动/手动轮椅510K,FDA注册,FDA美国代理人
5:ISO13485认证,CE评估报告编写,FDAQSR820等产品出口的相关认证
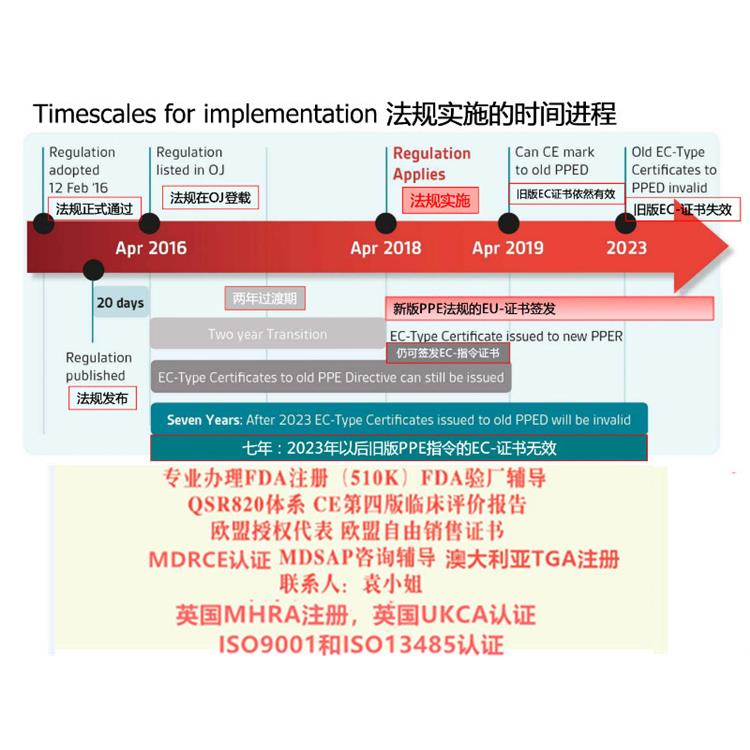
MDR实施之后,在三年过渡期内仍然可以按照MDD和AIMDD申请CE证书并保持证书的有效性。依据Article 120 clause2 的规定,过渡期内NB签发的CE证书继续有效,但是从其交付日期起有效期不超过5年,并且于2024年5月27日失效MDR已从指令升级为,这增强了其对欧盟成员国的约束力,并具有直接约束力。
加拿大MDEL注册 若您计划在加拿大销售器械,您需要进行产品注册登记以获得许可证加拿大颁发两种不同类型的许可证,两种许可分别有不同的要求。
新的器械法规(2017/745 / EU)(MDR)和体外诊断器械法规(2017/746 / EU)(IVDR)使欧盟立法与技术进步,科学变革和进步在法律制定上保持一致。新法规将建立一个健全,透明,可持续的框架,得到国际认可,可提高性并为制造商创造公平的市场准入。与指令不同,法规不需要转变为法。器械法规(MDR)的背景MDR将取代现有的器械指令(93/42/EEC)(MDD)和主动植入式器械指令(90/385/EEC) (AIMDD)。
我们该怎么办?
l 重新确认产品风险分类等级,确认是否有风险等级升级的情况?
例如部分可重复使用的器械,原属于ClassⅠ的器械,按照新法规变成了ClassⅠ*类器械。美容类产品原MDD下不属于范围,现MDR法规中已纳入;
l 确认原CE证的发证机构是否已获得欧盟当局批准的颁发MDR证的,目前拥有该的认证机构:BSI、TUV南德(注意TUV莱茵目前还未获得批准);
l 确认原CE认证时的技术文件中是否含有按照Rev4原则提供的评价报告;
l 确定企业合规负责人(MDR法规要求),有相应能力、和经验来承担相应的法规工作职责。
l 修改原CE技术文件,建立质量管理体系,向具有MDR发证的认证机构提出MDR-CE认证申请,获得MDR法规下的新CE证。
2017年2月Regulation (EU) 2017/745 on Medical Devices器械法规(MDR)提案发布,同年3月,欧盟成员国一致投票表决同意MDR。2017年5月5日,欧盟Official Journal正式对外宣MDR法规内容。
MDR新法规将取代现行的有源器械指令Council Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD) (1990)以及器械Council Directive 93/42/EEC on Medical Devices (MDD) (1993)指令。原计划2020年5月26日正式实施的MDR受全球影响将推迟实施时间至2021年5月26日。