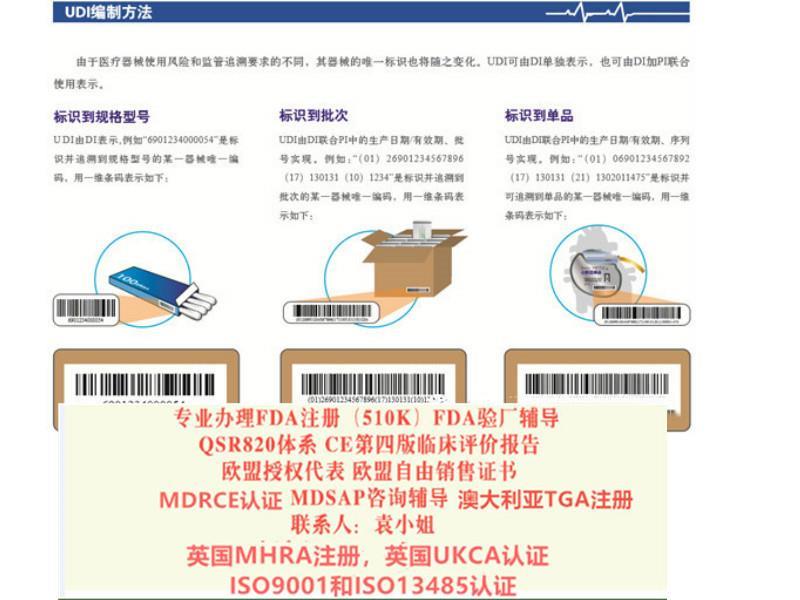
产品规格:
产品数量:
包装说明:
关 键 词:医疗器械FDA验厂费用
行 业:商务服务 认证服务
发布时间:2020-01-15
FDA简介
FDA隶属于美国健康与人类服务部(DHHS),负责全国品、食品、生物制品、化妆品、兽、器械以及诊断用品等的管理。
FDA检查法规依据
食品
FDA Food Safety Modernization Act (FA)
21 CFR 110 & 117 (CGMPs)
Hazard ysis & Critical ControlPoints (HACCP)
器械
21 CFR 820 (QSR820)
21 CFR 803 (MDR器械不良反应报告)
21CFR 801 (标签)
21 CFR 807 (510K注册)
品
21 CFR 210&211 (CGMP)
什么是FDA工厂检查
FDA派遣审核官员对食品/器械/品生产场所质量体系法规符合性进行检查.
FDA检查对象
适用于所有产品出口到美国的食品/器械/品企业
根据法规规定, 例行检查; (大多数情况,日常检查)
被FDA列到自动滞留的制造商,必须在FDA检查工厂检查通过后,才可以在美国市场上合法销售;(符合性跟踪检查)
产品在美国市场发生质量事故;(事故调查)
在与海关系统联网的数据显示制造商在某个时间器械产品进口数量非常大
产品在美国中了某些采购招标, 例如美国国防部
对于I类器械,约每四年检查一次;对于II/III类器械,约每两年检查一次。
FDA负责其审核官员的所有费用
FDA检查时间和
工厂属海外检查,FDA通常会提前2-3个月发通知
(因FDA在有办事处,且有常驻审核员,也可能会临时通知检查,如厦门森洋、江苏海江)
食品:1名检察官,1-2个工作日
器械:一类二类器械均为1名检察官;三类器械为1名或2名检察官,4-5个工作日
品:2名检察官(1名生物化学制背景、1名质量体系专家),4-5个工作日;
工厂检查的结果
FDA不发任何证书。
审核结束后现场会有以下两种情况:
无书面评价
收到书面的483表格
483表格用于执行检查的FDA检查官记录其所发现的缺陷。在检查结果时检查官要签发该表格,受检企业则需要对其中缺陷进行正式回复。对该表格中缺陷的回复需要在其签署后的15个工作日内提交。只有在收到回复后,FDA才会可能做出决定是否签发警告信。
除现场收到的483表格外,后续企业可能会收到:
Warning Letter 警告信(警告信一般在FDA上发布)
OAI引起
除了483表以外,检查官还要制作工厂调查报告EIR(Establishment Investigation Report)。EIR会在30个工作日内完成,然后交由FDA地区办公室或办公室负责人评审,评审后会被识别为以下几种状态:
NAI:不需要采取措施---在检查期间没有发现违规项目
VAI:自愿采取措施---发现了违规项目,但不需要采取行动。
OAI:需采取行动—发现违规项目,需要采取进一步法规措施
483回复引起
如果检查中发现了严重缺陷,且对483表格的回复被认为是不充分的,则FDA会签发警告信。
报价及周期
一般2-3个月,具体根据产品的风险等级、企业能力及配合程度有所不同。一般现场至少安排3次,每次至少2-3天:
次:差距分析,布置整改任务;
第二次:核对细节,检查整改情况;
第三次:全面审核,查漏补缺;
陪审。
FDA美国代理人服务/ FDA QSR820验厂辅导及整改/OTC验厂辅导及整改
FDA验厂咨询步骤首先做好提前准备检查接待,建立一个检查应对管理小组,内部审计项目,模拟检查项目。
-/gjhdff/-