欧盟医疗器械全新MDR法规和旧MDD指令的区别
价格:面议
专业办理欧盟CE认证(CE整套技术文件编订、 CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书
欧盟普通医疗器械条例EU 2017/745详解(上)
内容提要:
? 指令升级为条例
? 扩大监管的产品范围(附录XVⅠ)
? 补充了分级规则
? 减少并加严上市途径
? 修改了适用对象
? 修改了技术要求
? 新增了临床评价及调查要求
? 强化上市后要求,引入生命周期概念
? 强化质量体系要求
? 其他关注点
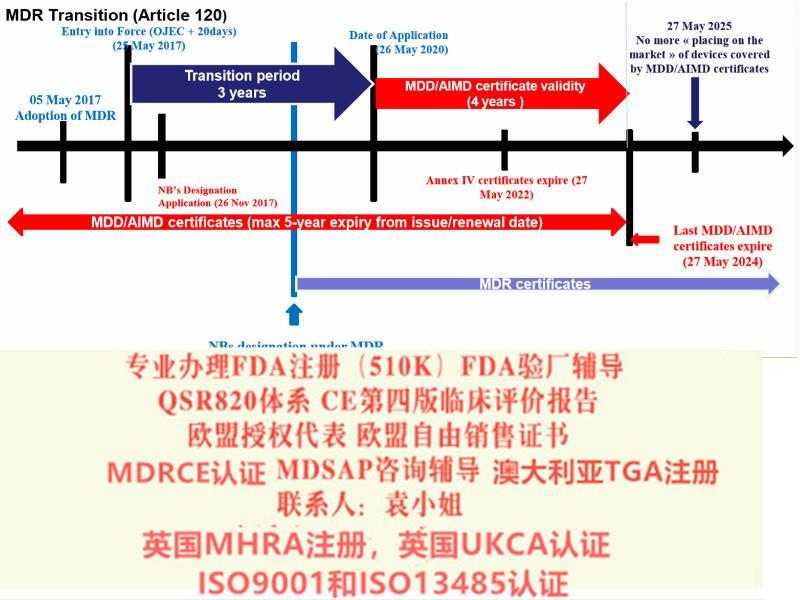
? 生效与过渡
一、指令升级为条例
? 90/385/EEC与93/42/EEC一起调整为EU 2017/745,有源植入器械指令与普通医疗器械指令一起,调整为普通医疗器械条例(MDD→MDR);
? 90/385/EEC和与93/42/EEC,必须经成员国转化为本国的法规才能产生作用, EU 2017/745无需成员国转化,即可直接生效;
? EU 2017/745还修改了其他指令,如2001/83/EC ;
二、监管的产品范围扩大
? 从使用目的上扩展:
---医疗器械清洁、消毒和灭菌的器械也是医疗器械(在13485:2003中已协调);
---支持受孕的器械;
---附录XVⅠ的器械(主要是美容类器械)
---体外诊断器械
(在13485:2003中已协调,这样,ⅠVDR不是一个与MDR并列的条例,也是MDR的补充)
? 从使用对象方面扩展:
专业用器械;非专业用器械;
? 从使用场所方面扩展:
上市销售的器械设备;不上市销售但投入使用的器械;
三、医疗器械分级
? 普通医疗器械,包括有源植入器械,但不包括体外诊断器械,共分四个风险等级,Ⅰ,Ⅱa, Ⅱb,Ⅲ级,Ⅰ级最低,Ⅲ级最高;
? 分级原则
---分级规则的运用应基于器械的预期用途;
---如果器械是与其它器械组合起来使用的, 分级规则应对每一器械分别运用. 附件应单独分级;
---软件, 驱动器械或改变器械的使用的, 与相应的器械同级.独立软件单独分级;
---不完全应用在人体特定部位的器械应依其最重要的用途来分级;
---当一个器械适用好几个分级规则时, 应根据制造商宣称的功能应用最高的分级;
---器械的持续作用时间应包括相同或同一器械的累积使用,如由于器械清洁消毒导致的器械临时中断使用或为更换另一同类器械而移走;
---当器械本身作出相关疾病或病情的诊断时,或能够提供诊断的决定性信息时,器械可被视为允许提供直接诊断。
四、符合性评定程序
?III级器械:
途径1:ANNEX IX完整质量体系。内容包括完整质量体系及监督审核,技术文件和上市后技术文件评审及特定产品的补充评审程序,文件保存;
途径2:ANNEX X型式检验+ANNEX XI生产质量保证或产品验证,生产质量保证或产品验证程序中包含技术文件的评审;
? IIb级器械:
途径1:ANNEX IX完整质量体系中的第I章、第III章及第II章中第4部分的技术文件评审。内容包括完整质量体系及监督审核,技术文件和上市后技术文件评审,文件保存;
途径2:ANNEX X型式检验+ANNEX XI生产质量保证或产品验证,生产质量保证或产品验证程序中包含技术文件的评审;
? IIa级器械:
途径1:ANNEX IX完整质量体系中的第I章、第III章及第II章中第4部分的技术文件评审。内容包括完整质量体系及监督审核,技术文件和上市后技术文件评审,文件保存;
途径2:ANNEX XI第10或第18规定的技术文件的评审;
? I级普通器械:
技术文件编写+符合性声明
? I级无菌、测量或重复使用的手术器械
途径1: ANNEX IX完整质量体系中的第I章、第III章。内容包括完整质量体系及监督审核,文件保存;
途径2: ANNEX XI PART A 生产质量保证;公告机构仅审核“无菌部分”、“计量学部分”、“清洁+消毒+灭菌+维护+说明书”;
? 含药器械:
途径1:上述程序之外,ANNEX IX第5.2部分,或
途径2:上述程序之外, ANNEX X第6部分(ANNEX 中的特定产品评审程序理解为与型式检验证书一致);
? 含有生物源性非活性物质的器械(或含有自人体孔道或皮肤吸收的物质的器械):
途径1:上述程序之外,ANNEX IX第5.3部分,或
途径2:上述程序之外, ANNEX X第6部分(ANNEX 中的特定产品评审程序理解为与型式检验证书一致);
普通定制器械:
ANNEX XIII
? III级植入式定制器械:
? 途径1: ANNEX XIII+ANNEX IX第I章全面质量体系;
? 途径2: ANNEX XIII+ANNEX XI PART A生产质量保证
五、供应链上,适用对象扩大
? EU 2017/745 不仅规定了制造商的职责,还专门规定了进口商和经销商的职责;
? 同时, EU 2017/745 还规定了欧盟代表的职责;
? EU 2017/745还辟出专门章节规定了公司机构的职责、资格认定要求、更换方法、工作流程等。
? 进口商和/或经销商,只要以自己的名义或品牌将产品引入欧盟市场,即需覆行制造商的职责,包括上市后监督职责;
? 对品牌商( PLM或OBL) ,EU 2017/745 要求完整的技术文件,这在现实在通常难以实现。这项要求,将使得逐渐消失;
?-/gbaaeee/-