框式助行器的MDR-CE认证 什么是MDRCE证书 申请要求
价格:面议
产品规格:
产品数量:
包装说明:
关 键 词:框式助行器的MDR-CE认证
行 业:商务服务 认证服务
发布时间:2023-11-25
如果制造商在欧洲没有实际的场地,需要任命一位欧洲授权代表,代表制造商在欧盟境内进行相关的法规联络事宜。
MDR对于DOC的要求
MDR 在其附录 IV中对DOC的内容作了明确的规定,至少包括如下内容:
制造商名称、注册商品名或注册商标和单一注册号(如签发)及其授权欧盟代表(如适用)和注册营业地点的联系地址
制造商对签发欧盟符合性声明负完全责任的声明
附录VI第C部分所所述的基本的器械标识UDI - DI
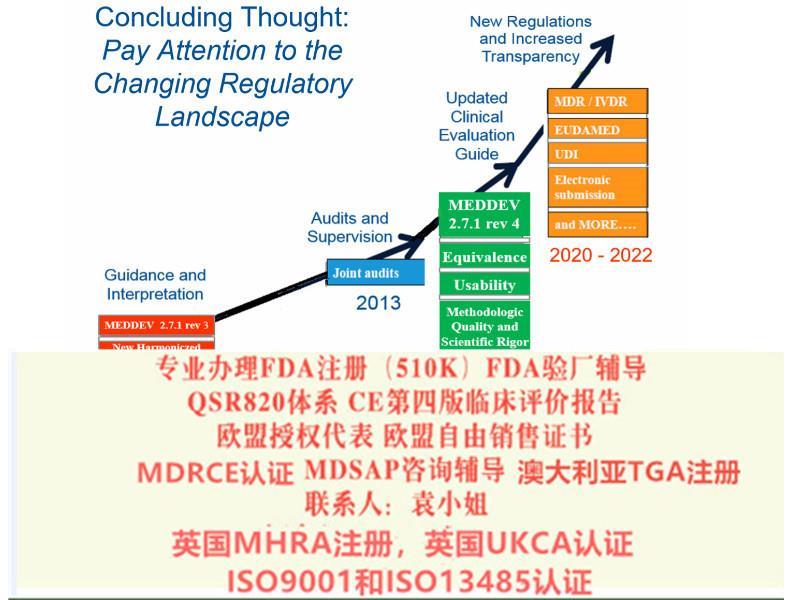
十二、完善评价相关要求
新法规提出:
要求根据Article61和附录XIV partA执行、评估、报告和更新评价资料;
提出对特定III类和IIb类器械,CER中要考虑咨询小组的意见;
对植入和III类器械,提出考虑研究;
要求CER按照PMCF取得数据进行更新;
针对III类和可植入器械,提出了CER更新的频率;
明确实质等同性需考虑的特点;
要求其与风险管理的相互作用
十三、Eudamed数据库
新法规提出:
明确欧洲器械数据库(Eudamed)建立目的和包含的信息(Article 33);
信息的公开性:
要求III类器械和植入式器械,和性能信息通过Eudamed向公众开放。
十四、提出器械的可追溯性(UDI)
除定制和研究器械外,其他器械均需建立UDI系统;
UDI信息体现在标签或包装上(不包含集装箱);
UDI-DI信息需要载明于符合性声明中(见Article27);
Annex VI Part B提出UDI-DI包含的信息;
可植入、重复使用、软件、可配置器械的UDI有要求(见Annex VI Part C)
包装或标签上UDI实施的时间见Article123 (f)。
UDI 发行实体由欧盟会。
过渡性:Article 120指出“在会根据第27(2)条发行实体前,GS1、HIBCC和ICCBBA应被视为的发行实体”。
MDR将取代Directives 90/385/EEC (有源植入类器械指令)and 93/42/EEC(器械指令)。依据MDR Article 123的要求,MDR于2017年5月26日正式生效,并与2020年5月26日期正式取代MDD(93/42/EEC)和AIMDD(90/385/EEC)。
说到这里 企业应该如何面对新法规的升级呢?按照MDR法规要求。关键的内容包括如下几个方面:
企业的质量管理体系 EN ISO13485:2016 ,产品的型式试验 TYPE TESTING , 产品的技术文件 TECHNICAL CONSTRUCTION FILES要满足这些要求,通常需要咨询机构和咨询师的协助。本公司拥有一支经验丰富的咨询师队伍可以为贵司提供这些服务,包括: 协助贵司建立/升级器械质量管理体系,将MDR法规的内容整合进去 协助贵司确定产品的欧盟协调标准,确认检测实验室的,样品准备以及检测不合格整改的研讨 按照MDR要求协助贵司准备技术文件,包括风险分析报告,评估资料,基本要求检查表等 协助贵司按照欧盟的要求修订说明书和标签,使其满足出口的要求。
我们可以为您提供的自主服务项目主要有: 出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售、防护服PPE指令Type5/6认证 出口美国法规:器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系、食品FDA验厂及整改、OTCFDA验厂及整改 中国法规:器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证、SFDA验厂、SFDA注册检测、企业标准编制、局自由销售证。