怎么办理欧盟mdr ce认证 ce属于什么认证 的流程及步骤
价格:面议
产品规格:
产品数量:
包装说明:
关 键 词:ce认证,怎么办理欧盟mdr
行 业:商务服务 认证服务
发布时间:2023-11-21
MDD指令和MDR法规的CE认证的区别
老MDD指令申请CE认证,由于法规规定产品在市场上出现任何问题,都是由制造商承担。其中欧盟授权代表的职责只是沟通协调以及产品包装可以使用欧盟授权代表的公司名字和地址信息的责任。国外的进口商更多的是找工厂要一张MDD的CE证书,能顺利清关销售便可以了,一般不关注你们这个证书怎么获得的,是否正真满足法规要求的。
但是新MDR的管控趋于严格,对于制造商,欧盟授权代表以及国外进口商三方该承担的责任比较明确,欧盟授权代表和进口商与制造商一样为缺陷器械承担连带的法律责任。所以进口商在采购工厂产品的时候,较MDD老法规,他们更关注,工厂是否真正满足CE法规要求,尽量的将自己要承担的风险降低到。
我们为企业编写的MDR CE技术文件里的:风险分析报告,评价报告,基本基本检查表等等,不仅仅是为了获得一张证书而做的,更多的都是从各个方面来产品是的有效的。
法规条款增加,认证评审更加严格
a. 分类规则增加:由MDD的18条增加到 22条;
b. 基本要求检查表条目增加:由MDD的13条增加到 MDR的23条;
c. CE技术文件的结构发生了变化,分为:产品技术文件和上市后文件(MDD只要求产品技术文件);
d.评价报告。MDR要求企业提供第四版评估报告,相比于第三版,第四版要求更为严格;
MDR要求更高的透明度和可追溯性
a. 引入了器械标识UDI,增加产品的可追溯性;
b. 企业的相关信息都会被收集到欧洲器械数据库(EUDAMED);
c. 建立上市后监督(PMS)系统;
出口美国需要的为:
FDA注册,FDA510K,QSR820体系(美国FDA验厂)
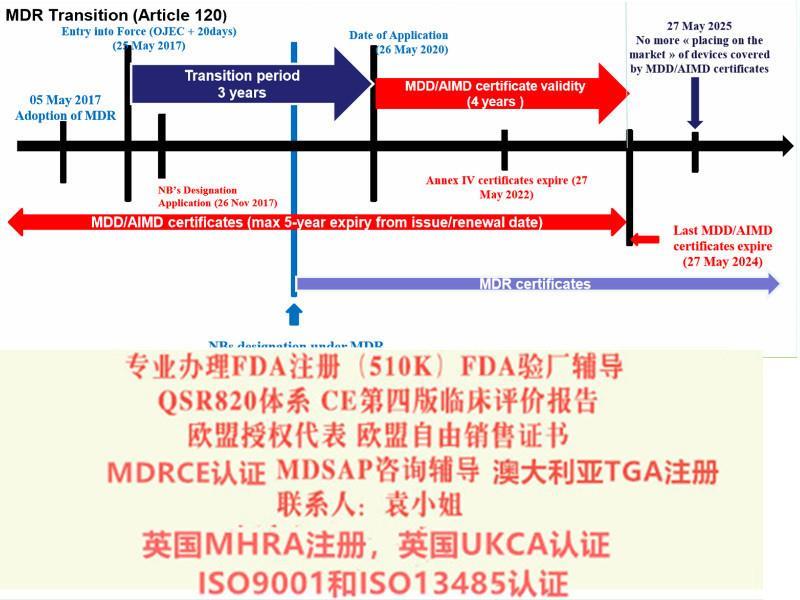
MDR 由指令升级为法规,提高了对欧盟成员国的约束力,具有直接约束性,无需各国转化为本国的法律法规的形式即可落实实施。内容上,MDR 在整合原指令的基础上,大幅提升了有关器械认证的规范和限制,例如关于产品分类规则、器械的可追溯性、性能研究的规范、增加上市后的产品性和有效性的等方面。MDR 共10 章123 条,并附有17 个附录。
在此基础上,会议对于MDR及IVDR的执行达成了如下时间表。2016年12月14日,EUDAMED(European databank for medical devices) 筹划会上欧盟各国对于器械法规MDR及IVDR的执行进行了一轮的讨论,与会人员对于这两个法规的细节内容进行了讨论并达成了一致意见。在此基础上,会议对于MDR及IVDR的执行达成了如下时间表。一,DMR的主要变化1.扩大了应用范围2.提出了新的概念和器械的定义3.细化了器械的分类4.完善了器械的通用和性能要求5.加强对技术文件的要求6.加强器械上市后的7.完善评价相关要求8.提出Eudamed数据库的建立和使用9.提出器械的可追溯性(UDI)10.对NB提出严格的要求MDR简介2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。MDR将取代Directives 90/385/EEC (有源植入类器械指令)and 93/42/EEC(器械指令)。
IIa、IIb和III类器械制造商应针对各器械或类别或器械组编制定期性更新报告(PSUR),总结根据上市后计划收集的数据分析结果和结论,并对采取的任何预防和纠正措施提供理由和说明。
IIa类器械制造商应在必要时至少每两年更新PSUR ,IIb和III类器械的制造商应至少每年更新PSUR。
警戒 (MDR第87~92条)
瑞士也已经不认可欧洲的CE认证,您有产品出口瑞士吗?是否有做瑞士代表以及瑞士注册的?