儿童体温贴EU2017/745认证介绍 全系列解决方案
价格:面议
MDR欧盟授权代表和MDD欧盟授权代表的区别: 欧盟授权代表(AR)是指在欧盟境内的任何自然人或法人,其收到并接受位于欧盟以外的制造商的书面授权,代表该制造商按照本法规对制造商所规定的义务要求所进行的一切行动。
什么是MDRCE证书,编写MDRCE技术文件需要多少钱
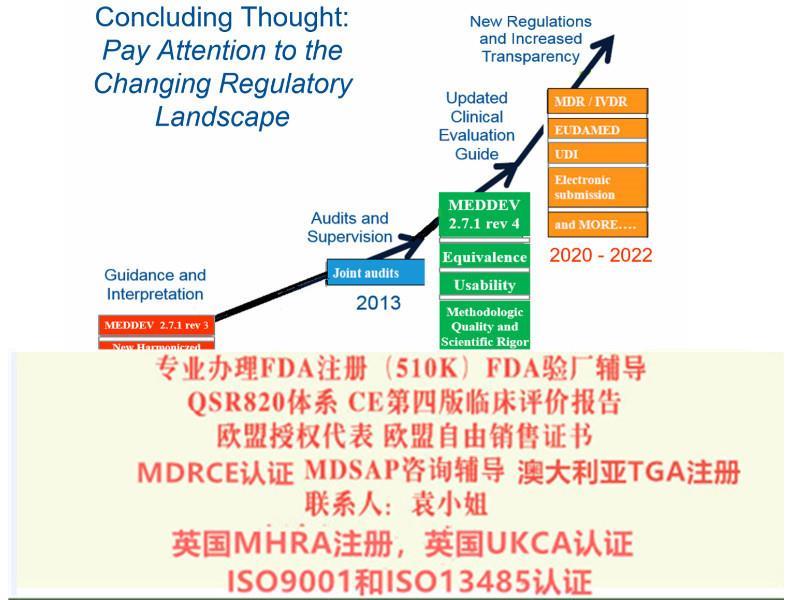
欧盟第四版评价(MEDDEV 2.7.1 Rev 4)指南主要变化a)报告更新的频率b)报告编写人和评价人的c)评价报告需要有明确的可测量的目标d)确定技术发展水平e)数据的科学性和有效性f)比对器械g)比对器械的数据获得h)什么时候需要试验i)风险-收益j)售后监督和售后跟踪8)提出Eudamed数据库的建立和使用9)提出器械的可追溯性(UDI)10)对NB提出严格的要求2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。MDR将取代Directives 90/385/EEC (有源植入类器械指令)and 93/42/EEC(器械指令)。
关于器械编写MDR的CE技术文件需要多少钱?
MDR实施之后,在三年过渡期内仍然可以按照MDD和AIMDD申请CE证书并保持证书的有效性。依据Article 120 clause2 的规定,过渡期内NB签发的CE证书继续有效,但是从其交付日期起有效期不超过5年,并且于2024年5月27日失效
MDR已从指令升级为,这增强了其对欧盟成员国的约束力,并具有直接约束力。无需各国转换为国家法律法规即可实施。在内容方面,MDR基于原始指令的整合,大大改进了医疗器械认证的规范和限制,例如产品分类规则,设备可追溯性,性能研究规范,上市后产品安全性提高以及有效性方面和。 MDR由10章和123篇文章组成,共17个附录。
关于的过渡期:
仅拥有根据指令90/385/EEC和93/42/EEC颁发的证书的设备可以投放市场,前提是自MDR应用之日起设计和预期用途没有发生重大变化且符合要求新规定。,市场监督,警报,经济运营和设备登记的规定。
可通过免税订单获得,符合新规定的设备可在2020年5月26日前投放市场。符合新规定的合格评定机构可在2020年5月26日前并通知。可以采用合规合规评估程序,并在2020年5月26日之前根据新规定颁发证书。
我公司申请:出口瑞士:需要瑞士代表,瑞士注册