固定支具出口2017/745认证要求 全系列解决方案
价格:面议
产品规格:
产品数量:
包装说明:
关 键 词:固定支具出口2017/745认证要求
行 业:咨询 管理咨询
发布时间:2023-09-18
对制造商和产品的影响而言,93/42 EEC指令和MDR基本上具有相同的基本要求。没有现有的需求,但是MDR添加了新的需求,与目前93/42指令相比,MDR更加强调生命周期方法的性,并有数据支持。MDR对公告机构的提出了更严格的要求,对主管当局和会加强了控制和监测。
谁能做MDRCE技术文件,需要什么资料?
合格评定根据某些装置的危险类别和具体特征,对符合CE标志的装置的评价各不相同(第五十二条)。所有IIa、IIb和III类设备以及一些特定的I类设备都需要一个通知机构的干预(见第7a5、b6和c7段)。第52条和附件九、十、习叙述了根据设备类别不同的评估方法。在某些情况下,制造商对合格评定路线有一些选择。对于某些III类和IIb类设备,有一个新的评估咨询程序,该程序将由一个立的组根据该公告机构的评估评估报告进行(第五十四条)。附件一规定了一般的和性能要求,附件二和三规定了技术文件的组成。质量管理体系的范围(第10条第9款)包括评价和上市后随访。评价计划必须先于评价本身(附件十四,A部分)。
CEMDR认证-CEMDR认证和MDRCE认证的区别
2017 年4 月5 日,欧洲议会和理事会正式签发了欧盟关于器械第2017/745 号法规(MDR,EU2017/745),5月5日,欧盟(Official Journal of the EuropeanUnion) 正式发布该法规。2017 年5 月25 日,MDR 正式生效, 替代了原器械指令(MDD,93/42/EEC)和主动植入式器械指令(AIMD,90/385/EEC)。
MDR 由指令升级为法规,提高了对欧盟成员国的约束力,具有直接约束性,无需各国转化为本国的法律法规的形式即可落实实施。内容上,MDR 在整合原指令的基础上,大幅提升了有关医疗器械认证的规范和限制,例如关于产品分类规则、器械的可追溯性、性能研究的规范、增加上市后的产品安全性和有效性的等方面。MDR 共10 章123 条,并附有17 个附录。
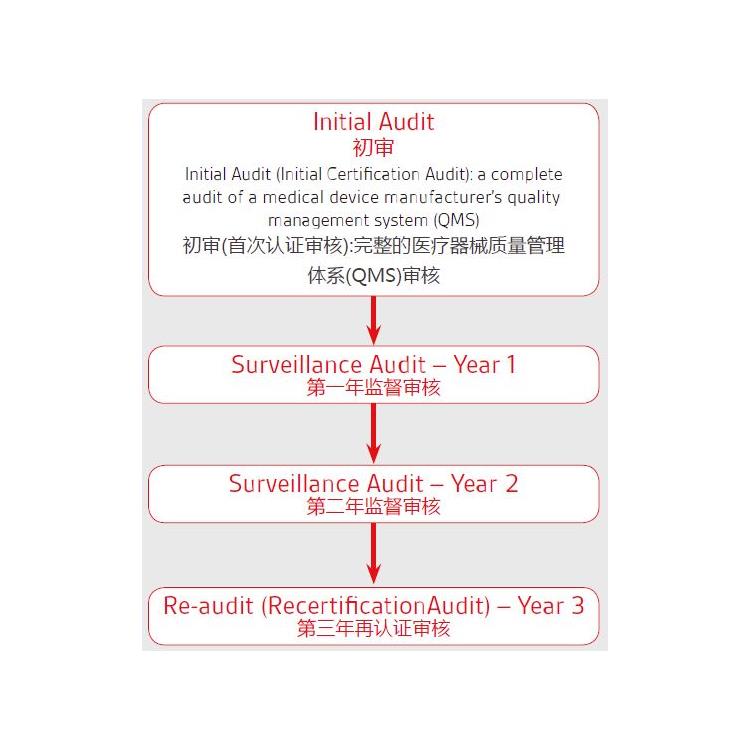
一、关于法规过渡期
MDR 过渡期为3 年,共涉及四个时间点(见表1):
欧盟器械新法规MDR主要变化情况介绍
仅具有根据90/385/EEC 和93/42/EEC 指令签发的证书的器械可投放市场的前提是自MDR 适用之日起,其在设计和预期目的上无显著变化并符合新法规有关市场后监察、市场监察、警戒、经济运营商及器械注册的规定。
通过豁免指令的形式上市,且符合新法规的器械可在2020 年5 月26 日之前投放市场。并可在2020 年5 月26 日前并通知符合新法规的符合性评估机构。公告机构可在2020 年5 月26 日前, 采用合规的符合性评估流程并按照新法规签发证书。
英国已经脱欧了,不再认可欧洲的CE证书,需要UKCA认证,有英代,MHRA注册,才可以合规出口英国。