体外诊断产品的FDA510K认证的周期多久 510K认证
价格:面议
我公司办理欧盟,美国,澳大利亚以及中东南美等等国家各类认证:FDA510K认证,欧盟自由销售证书,欧盟授权代表,ISO13485/ISO9001认证,欧盟CE认证(MDR(REGULATION (EU) 2019/745)),FDA注册,FDA验厂,英国授权代表,MHRA注册,美国代理人服务,澳大利亚TGA认证,CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、防护服PPE指令Type5/6认证、器械单一体系审核MDSAP认证、BSCI验厂、BRC 认证,澳大利亚TGA注册、口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试,器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证
美国食品和药物管理局( FDA )简介:
FDA 是食品药品监督管理局( Food and Drug Administration )的简称。FDA通常代表也代表美国 FDA ,即美国食品药品监督管理局,美国 FDA 是国际审核机构,由美国国会即联邦授权,从事食品与药品管理的高执法机关;是一个由、律师、微生物学家、药理学家、化学家和统计学家等人士组成的致力于保护、促进和提高国民健康的卫生管制的机构。全球其他许多国家也会通过寻求和接收FDA的帮助来促进并其本国产品的安全。
美国FDA是美国在健康与人类服务部(DHHS)下属的公共(PHS)中设立的执行机构之一。隶属于美国卫生教育福利部,负责全国药品、食品、生物制品、化妆品、兽药、
器械以及诊断用品等的管理。总部设在华盛顿特区及马 利兰州罗克威尔城,机构
庞大,分支机构遍布全国各地。FDA主要分测试和注册两个内容,器械化妆品食品药品类产品需要进行 FDA 注册, FDA 注册可以直接在FDA上进行申请。
我公司办理欧盟,美国,澳大利亚以及中东南美等等国家各类认证:FDA510K认证,欧盟自由销售证书,欧盟授权代表,ISO13485/ISO9001认证,欧盟CE认证(MDR(REGULATION (EU) 2019/745)),FDA注册,FDA验厂,英国授权代表,MHRA注册,美国代理人服务,澳大利亚TGA认证,CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、防护服PPE指令Type5/6认证、器械单一体系审核MDSAP认证、BSCI验厂、BRC 认证,澳大利亚TGA注册、口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试,器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证
基于的行为,必须向FDA递交510(k)的如下所示:
1) 把器械引入美国市场的国内厂家;
如果成品器械厂家根据他们自己的规范装配器械,并在美国上市,那么必须递交510(k)。然而,器械组件厂家并不要求递交510(k),除非这些组件销售给终用户作为替换零件。合同厂家,这些公司根据其他的规范按照合同装配器械,不要求递交510(k)。
2) 把器械引入美国市场的规范制订者;
FDA审查规范制订者与审查厂家几乎一样。规范制订者是制订成品器械规范的人,但是器械按照合同由其他的公司来生产。因此,规范的制订者,而不是合同厂家需要递交510(k)。
3) 改变标注或操作严重影响器械的再包装者或再标注者;
如果再包装者或再标注者严重改变了标注或影响了器械的其他条件,可能会要求递交上市前通知书。此时,你必须确定是否通过修改指南,或增加了警告,禁忌征候等等而显著改变了标注,还有包装操作是否能够改变器械的条件。然而,大多数的再包装者或再标注者并不要求递交510(k)。
4) 把器械引入美国市场的外国厂家/出口商或外国厂家/出口商的美国代理方。
510(k)规范(21 CFR 807)对于使用目的的主要变化,特别要求递交上市前通知书
我公司办理欧盟,美国,澳大利亚以及中东南美等等国家各类认证:FDA510K认证,欧盟自由销售证书,欧盟授权代表,ISO13485/ISO9001认证,欧盟CE认证(MDR(REGULATION (EU) 2019/745)),FDA注册,FDA验厂,英国授权代表,MHRA注册,美国代理人服务,澳大利亚TGA认证,CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、防护服PPE指令Type5/6认证、器械单一体系审核MDSAP认证、BSCI验厂、BRC 认证,澳大利亚TGA注册、口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试,器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证
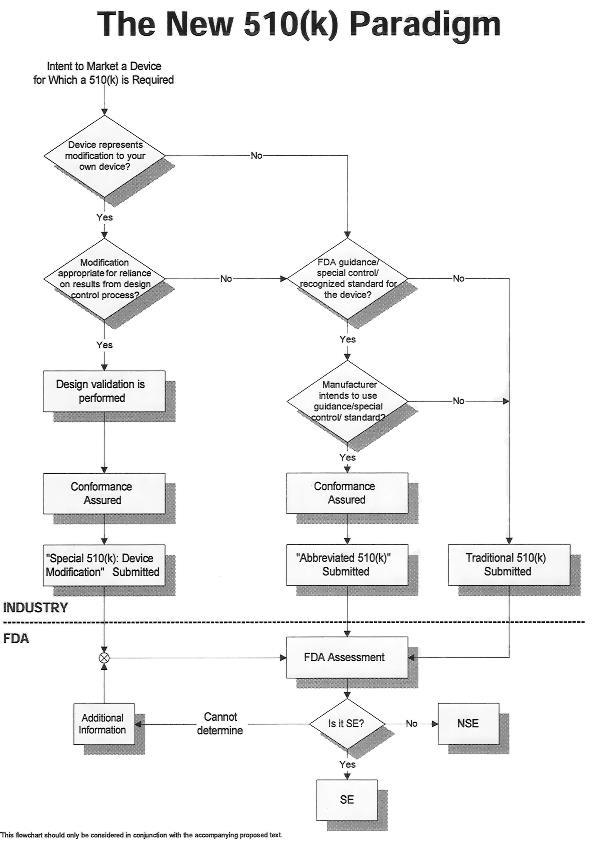
FDA 510K基本流程
美国FDA对流入本国并在本国上市销售的器械产品,实行严格的法规把控,510K产品在申报时必须通过FDA审查。
510K批准大致流程如下:
1 确定产品分类;
2 确定比对器械;
3 进行产品测试;
4 申报资料的提交、登入、审查与整改;
5 FDA评审批准。
PS:申报510K的企业同样需要进行企业登记和产品列名。