天津医疗器械单一体系审核MDSAP辅导
价格:面议
我们的服务:协助您按照MDSAP建立质量体系,协助您在原有的质量体系上符合MDSAP的要求
医疗器械单一体系审核MDSAP认证咨询辅导
(1)WHAT
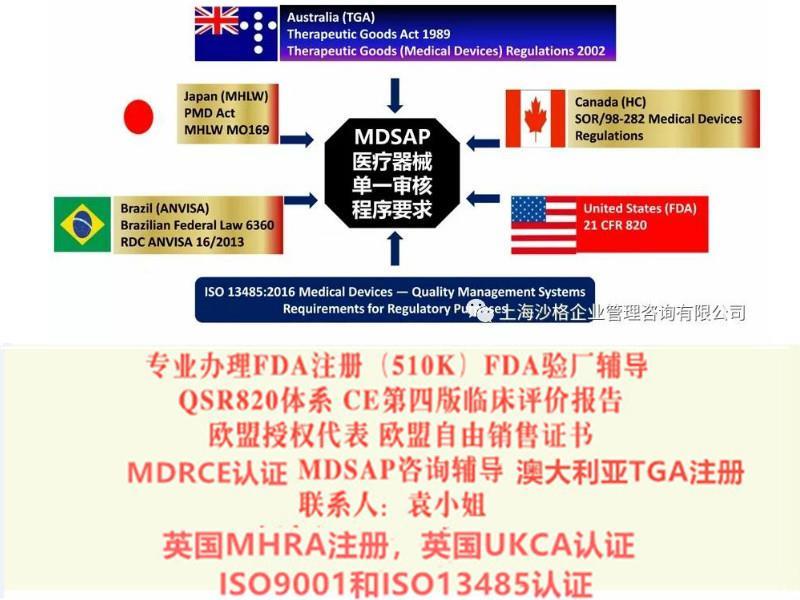
医疗器械单一审核程序”(Medical Device Single Audit Program (MDSAP) )项目是美国(FDA)、澳大利亚(TGA)、巴西(ANVISA)、加拿大(HC)、日本(MHLW)五国的监管机构认可并加入的一套新的审核程序。该程序旨在建立一套单一审核的过程,满足并统一上述国家的审核要求,使审核更加全面有效。以上五国监管机构认可MDSAP的审核结果,生产企业可以减少对应不同监管机构的审核次数,减少因审核带来的生产干扰。MDSAP的出台,使审核过程国际标准化,减轻了生产企业的负担。ISO13485与MDSAP的对比:ISO13485是MDSAP的基础;MDSAP的要求要高于/多于ISO13485的要求;ISO13485是标准,MDSAP除了ISO13485之外,还有各参与国的法规要求;两种认证,核心都是质量管理体系。
(2)WHY
根据加拿大卫生局已宣告的MDSAP过渡计划,CMDCAS证书(我司现有的ISO 13485证书)在2019年12月31日后将不被接受。为了继续持有现存的医疗器械许可证,制造商将在2019年1月1日前被要求提交有效的MDSAP质量管理体系证书。MDSAP加拿大过渡计划:加拿大卫生局打算将MDSAP实施列为制造商证明其符合质量管理体系要求的唯一途径。MDSAP于2019年1月1日开始实施过渡,过渡期两年。在这两年内,加拿大卫生局同时接受CMDCAS和MDSAP证书。2019年1月1日起,加拿大卫生局只接受MDSAP证书,CMDCAS质量管理体系证书需要被MDSAP质量管理体系证书替代。
(3)WHO
MDSAP由医疗器械生产企业联系第三方公告机构(如:SGS、BSI、TUV)进行审核,参与国(五国:美国、澳大利亚、巴西、加拿大、日本)的医疗器械监管部门可以将此审核报告作为判断依据。相关的程度如下:美国:替代FDA的常规检查(FDA专项和PMA产品除外);巴西:对于三类和四类医疗器械,可以替代ANVISA的上市前GMP检查,以及上市后的例行检查(专项检查除外);日本:对于II类,III类,IV类医疗器械,可豁免现场工厂审核;加拿大:2019年起强制取代CMDCAS,作为分类在II类及以上产品进入加拿大的唯一途径;澳大利亚:可豁免TGA审核,支持颁发和保持TGA符合性审核证书
(4)WHEN
按照加拿大过渡期计划,为了在强制日期(2019年1月1日)前取得MDSAP证书,考虑到公告机构的审核时限及证书审核和出具周期(约2个月),审核公司建议我司在2019年年初预约审核,建议在2019年6月之前完成现场审核。(注:公告机构约15天完成一个公司的现场审核及整改情况审核,沪X齿科和山东X鸽两个公司分开申请,合计30天)
(5)WHERE
由公告机构到企业进行现场审核。
(6)HOW
第一步,企业识别MDSAP法规,由质量管理部按照MDSAP的要求进行内审;第二步,由公告机构到企业进行现场审核;第三步,企业根据公告机构审核提出的整改项进行整改,15天内提交整改计划;第四步,公告机构审核企业的整改情况;第五步,审核通过,公告机构内部走出证审批程序,颁发证书。
(7)HOW MUCH
审核公司决定终的报价,目前CTS综合几家在中国开展认证业务的公司预估的费用如下: a) 初次审核:(约11-19万) b) 年审:(约6.6-9万)。
医疗器械单一体系审核MDSAP认证咨询辅导
1、USA FDA美国
FDA将接受 MDSAP试点审核报告用于代替 FDA例行视察(两年一次)。FDA将不会接受 MDSAP检查用于替代首次审核或“ “for cause ”或者“Compliance Follow-up”的审核。
2、Canada加拿大
从2019年1月1号至2019年1月1号,加拿大卫生部会接受按照CMDCAS和MDSAP要求颁发的证书,但是从2019年1月1号起,将只接受MDSAP证书。
3、ANVISA巴西
ANVISA对于申请在巴西上市的III类或者IV类产品的GMP证书申请,可能会用MDSAP试点审核代替由ANVISA进行的上市前审核。ANVISA也可以使用MDSAP试点审核来代替ANVISA的全面检查,用于申请每两年需要更新的GMP证书。
4、TGA澳大利亚
TGA在评估是否颁发或者维持法规规定的产品制造商的合格评定证书时,将会考虑MDSAP试点审核报告。在某些情况下,制造商可免于TGA的例行审核。当ISO13485:二003用于证实制造商符合澳大利亚合格评定程序的部分要求时,TGA将接受MDSAP证书作为符合此标准的证据。
5、MHLW & PMDA日本
日本厚生省和日本制药和医疗设备局将在日本按照法规要求上市前和定期的上市后监督审核中使用MDSAP审核报告代替现场J-QMS审核。
● 医疗器械行业从业16年经验,医疗器械法规、体系咨询师
● 精通医疗器械CFDA,FDA,CE法规
● 熟练掌握ISO13485/YYT 0287, ISO14971/YYT 0316, QSR820, JGMP, GMP 质量体系等
● 擅长美国FDA QSR820医疗器械辅导,熟练掌握QSR820各难点子系统的要求
● 具有几十家医疗器械企业咨询辅导成功经验和案例,帮助企业顺利通过美国FDA QSR820的审核,并部分企业获得零缺陷通过;并辅导企业获得ISO13485证书及CE证书,CMDCAS证书等。主要辅导的企业包括:新华医疗,怡和嘉业,河北普康等
●对于美国法规有经验丰富的专业团队,能够及时高效的提供服务。目前我们是国内唯一一家成功提供了FDA警告信解除和黑名单移除辅导的咨询机构
医疗器械单一体系审核MDSAP认证咨询辅导
医疗器械单一审核程序(MDSAP)是由国际医疗器械监管机构论坛(IMDRF)的成员共同发起的项目。旨在由具有资质的第三方审核机构,对医疗器械生产商进行一次审核即可满足参与国不同的QMS/GMP要求。
五家监管机构(澳大利亚医疗用品管理局,巴西国家卫生监督局,加拿大卫生部,美国食品和药品管理局,日本厚生劳动省和医药医疗器械管理机构)已正式加入MDSAP三年的试点计划。世界卫生组织以观察员的身份正式加入。BSI已获该审核机构资质,正全力支持和推进MDSAP试点计划。
参与 MDSAP 试点的监管机构
MDSAP试点计划已有显着进展,以下是参与试点的五国监管机构就如何使用MDSAP报告的声明:
澳大利亚:医疗用品管理局(TGA)将使用MDSAP的审核报告作为评估符合医疗器械市场批准要求的部分证据 ,除非该医疗器械可以豁免上市批准要求,或者当前的法规不允许使用MDSAP的报告。
巴西:国家卫生监督局(ANVISA)将采用MDSAP审核结果以及审核报告 作为产品上市前和上市后审核程序的重要输入,适当时,一些关键的信息还可以支持法规的技术评审 。对于III 或IV类医疗器械,ANVISA可能使用MDSAP审核代替这些产品在巴西上市前的GMP审核。GMP是上市的必要条件,通过MDSAP审核可能会加速GMP认证过程。
加拿大:加拿大卫生部在3年的试点期间, 同时认可MDSAP审核和加拿大医疗器械符合性评估系统(CMDCAS)审核。依据加拿大医疗器械法规第32章,MDSAP证书或CMDCAS证书都可被加拿大卫生部接受,用来申请新的(或维持现有)II,III或IV类医疗器械注册证。
美国:美国食品和药品管理局的器械和放射健康中心(FDA CDRH)将接受MDSAP审核报告替代FDA例行检查。FDA开展的“有因检查”或“合规跟进检查”不会受此计划影响。 此外,MDSAP不适用于上市前批准(PMA)的器械所必须的上市前审核和上市后审核,以及法案(21 U.S.C.30c (f)(5))中513(f)(5) 章节下关于产品分类界定的器械。
日本:日本厚生劳动省 (MHLW)以及医药医疗器械管理局(PMDA)对在日本法规框架内的产品上市前和上市后的定期审核 都将使用MDSAP的审核报告。
MDSAP 的优势
通过MDSAP审核将:
? 减轻医疗器械制造企业多重法规审核的负担
? 提供可预测的审核计划(包括审核开始和结束日程)
? 利于患者的健康和了解(消除工厂审核的障碍)
? 优化监管资源配置
? 可与ISO 13485和CE符合性审核相结合
? 多国法规要求一次审核完成
美国,澳大利亚,巴西,加拿大,日本五国的监管机构,加入“医疗器械单一审核程序(Medical Device Single Audit Program (MDSA))”项目并试行。该程序旨在建立一套单一审核的过程,满足并统一上述国家的审核要求,使审核更加全面有效。
2015年1月1日,在全球范围内,对于美国,澳大利亚,巴西,加拿大市场感兴趣的生产企业受邀参加了该项目。去年夏天,日本正式成为MDSAP的一员,对于日本市场感兴趣的生产企业也将受到该项邀请。
MDSAP由第三方进行审核,参与国的医疗器械监管部门可以将此审核报告作为判断依据。MDSAP不仅减少了监管部门例行检查的工作,也使得各监督部门共享相同且可靠的审核结果。
由于监管机构认可MDSAP的审核结果,生产企业可以减少对应不同监管机构的审核次数,减少因审核带来的生产干扰。MDSAP的出台,使审核过程国际标准化,减轻了生产企业的负担。MDSAP的审核依照参与国的已有法规要求,不额外增加其他要求,绝大多数情况下,各国的要求采用的是协调标准或标准内容基本类似,例如:医疗器械质量管理体系用于法规的要求(ISO 13485:二016),巴西的良好生产规范(RDC ANVISA 16/2013),美国质量体系规范 (21 CFR Part 820)和MDSAP参与国的其他特定上市前和上市后的法规要求。