医用检查手套CE认证 SUNGO的欧代 欧盟授权代表操作
价格:0.00起
产品规格:
产品数量:
包装说明:
关 键 词:医用检查手套CE认证
行 业:咨询 管理咨询
发布时间:2023-12-19
我公司办理:出口美国需要FDA注册,FDA510K,美国代理人(SUNGO可以做510K以及满足FDA要求的510K检测报告,7月份签约价格有优惠)
MDRCE认证,CEMDR认证,编写CE技术文件,CE*四版评价报告,(MDRCE技术文件:Medical Device Regulation 2017/745/EU) 我公司是做: 新MDR法规和MDD指令的欧盟CE认证,CE*四版评价报告更新/编写 欧盟自由销售证书,欧盟授权代表(德国,荷兰,英国), ISO9001/ISO13485认证以及咨询 FDA注册,FDA510K,FDA验厂/陪审和翻译 MDSAP咨询,国内注册证,生产许可证的办理

按照附录VIII的规则所划分的器械风险等级 当前声明所涵盖的器械符合本法规,以及适用时其他相关的要求签署欧盟符合性声明的欧盟立法的声明 符合性声明中所用的任何通用规范的索引公告机构的名称和标识号(如适用),所执行的符合性评估程序的说明和所签发的证书的标识 如适用,额外的信息 签字人的声明,地址和日期、签字人姓名和职务、以及代签人签名

在此基础上,会议对于MDR及IVDR的执行达成了如下时间表。2016年12月14日,EUDAMED(European databank for medical devices) 筹划会上欧盟各国对于器械法规MDR及IVDR的执行进行了一轮的讨论,与会人员对于这两个法规的细节内容进行了讨论并达成了一致意见。在此基础上,会议对于MDR及IVDR的执行达成了如下时间表。一,DMR的主要变化1.扩大了应用范围2.提出了新的概念和器械的定义3.细化了器械的分类4.完善了器械的通用和性能要求5.加强对技术文件的要求6.加强器械上市后的7.完善评价相关要求8.提出Eudamed数据库的建立和使用9.提出器械的可追溯性(UDI)10.对NB提出严格的要求MDR简介2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。MDR将取代Directives 90/385/EEC (有源植入类器械指令)and 93/42/EEC(器械指令)。
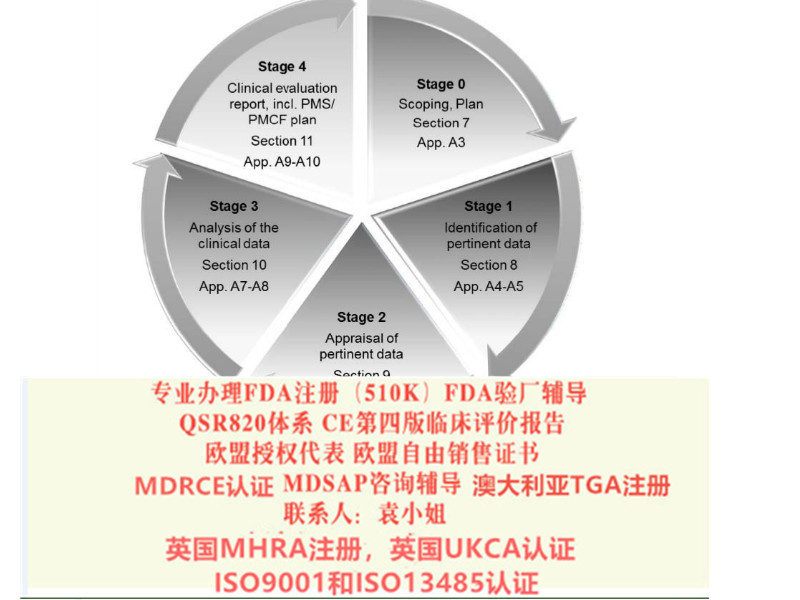
出口美国需要的为: FDA注册,FDA510K,QSR820体系(美国FDA验厂) 出口非欧盟为: 国外的客户,想要进口中国工厂的产品,需要客户,先要把产品在当地的局进行注册,完成注册后,才可以进口,销售。
法规条款增加,认证评审更加严格, CE技术文件的结构发生了变化,分为:产品技术文件和上市后文件(MDD只要求产品技术文件);


