全自动生化分析仪MHRA注册要求 英国代表是什么
价格:0.00起
产品规格:
产品数量:
包装说明:
关 键 词:全自动生化分析仪MHRA注册要求
行 业:咨询 管理咨询
发布时间:2023-12-02
SUNGO所有客户都有一对一的客服对接以保持经常性的联系,提供在线即时服务,针对贸易中存在的任何技术壁垒方面的问题提供的支持和解。
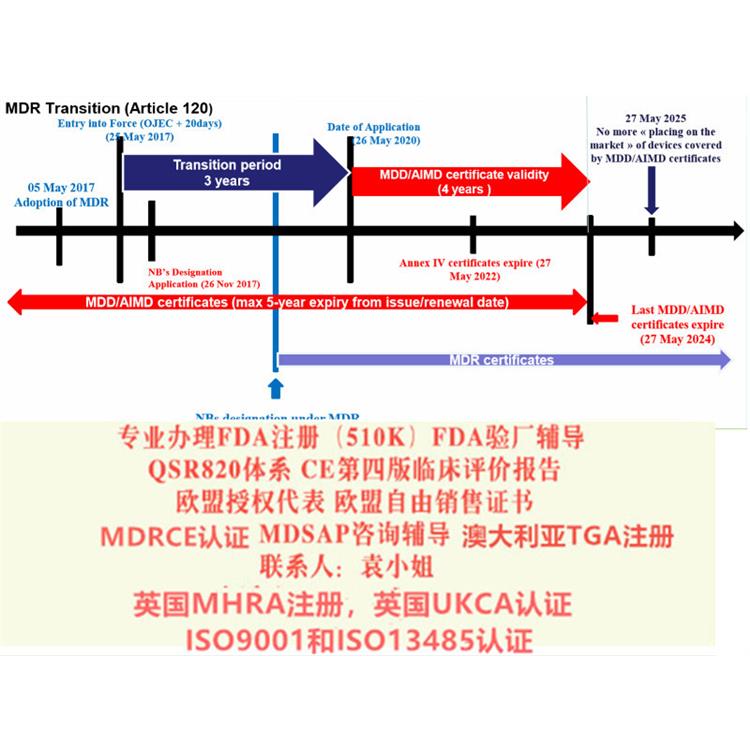
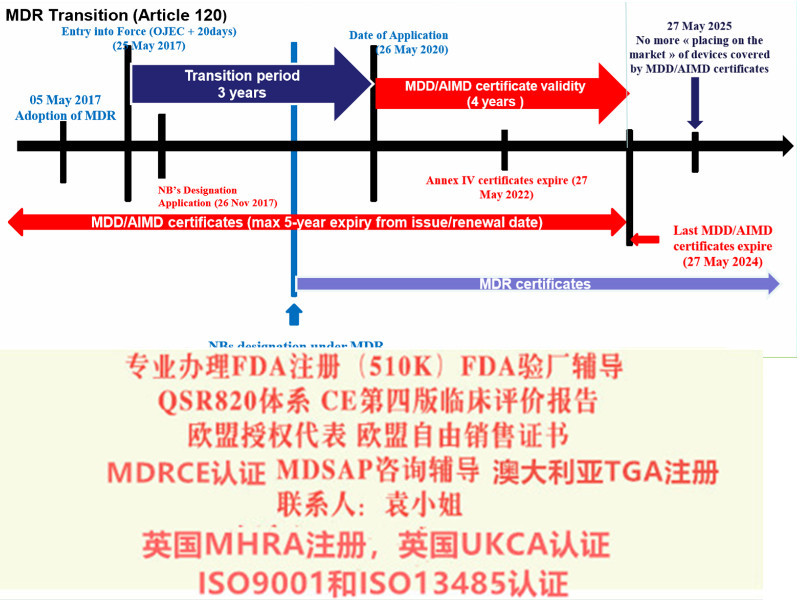
给大家带来解读MDR系列讨论 部分:演变过程和MDR的过渡期。众所周知,现行的MDD 器械指令93/42/EEC是1993颁布的,距今已经有26年的历史,这期间,器械行业无论是从技术方面,应用方面都有了巨大的变革,无疑,一部用了26年的指令已经过于陈旧,新法规替代老法规已经势在必行。2010年发生的法国PIP事件也促使了欧盟推行欧盟新法规的起草和推行。
EAR应帮助制造商履行哪些职责 依据欧盟法规,EAR承担相应职责包括以下内容: 1.法律职责和具体内容 1)通知主管当局制造商地址; 2)通知主管当局制造商的产品; 3)通知主管当局产品的变更; 4)向主管当局通报表现特征; 5)起草有关设备性能评估的声明; 6)**条款中作为欧盟会的联系方; 7)可启动合格评定程序; 8)根据主管当局的要求向其提供技术文件; 9)接受主管当局事件的通知; 10)公告机构和制造商之间的接口; 11)如果错误地贴上CE标志,授权代表必须终止侵权行为; 12)对于用于研究的设备,授权代表应遵循规定的程序并通知主管当局。

2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。

管理条例规定的职责和具体内容 1)代表制造商; 2)应要求向主管当局提供制造商授权委托其为EAR的副本; 3)验证制造商起草的欧盟符合性声明和技术文件; 4)在适用的情况下,验证制造商是否已执行适当的合格评定程序; 5)保留一份技术文件、符合性声明的副本,如果适用,还应保留一份相关证书的副本,供主管当局使用; 6)遵守注册义务; 7)验证制造商设备注册所需承担义务的符合性; 8)应要求向主管当局提供必要的信息和文件,以设备的一致性; 9)向制造商发送主管当局对样品或设备访问的任何请求,并验证主管当局是否收到样品或获得设备访问权限; 10)与主管当局合作,采取任何预防或纠正措施,以或减轻设备造成的风险; 11)向制造商通报人员、患者和用户对其设备相关疑似事件的投诉和报告; 12)应在与制造商相同的基础上对有缺陷的设备承担法律责任,并与制造商承担连带责任。 [img]http://img.qy6.com/new19/sungo99/1565589799.jpg[/img] (三)如何选择EAR
例如,MDR明确涵盖用于清洁、或其他设备的所有装置(*2.1条);一次性器械(*17条)};和某些无用途的装置(附件十六)。MDR还包括在互联网上销售器械以及用于远程提供诊断或服务的器械(*6条)。MDR为一些Ilb类器械和植入性Il类器械引入了由立小组进行的评估咨询程序(*54条)。