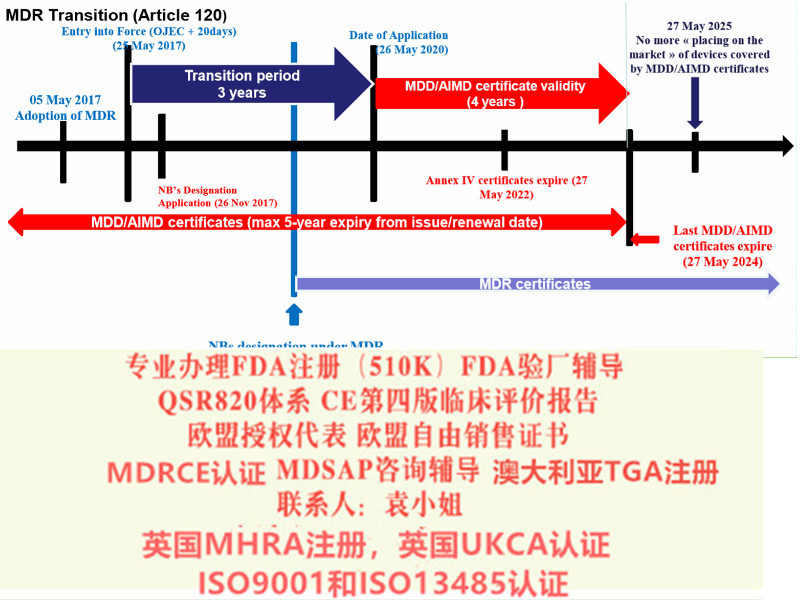
我们该怎么办? l 重新确认产品风险分类等级,确认是否有风险等级升级的情况? 例如部分可重复使用的器械,原属于ClassⅠ的器械,按照新法规变成了ClassⅠ类器械。美容类产品原MDD下不属于范围,现MDR法规中已纳入; l 确认原CE证书的发证机构是否已获得欧盟当局批准的颁发MDR证书的资质,目前拥有该资质的认证机构:BSI、TUV南德(注意TUV莱茵目前还未获得批准); l 确认原CE认证时的技术文件中是否含有按照Rev4原则提供的评价报告; l 确定企业合规负责人(MDR法规要求),有相应能力、资质和经验来承担相应的法规工作职责。 l 修改原CE技术文件,建立质量管理体系,向具有MDR发证资质的认证机构提出MDR-CE认证申请,获得MDR法规下的新CE证书。 Q:.对于QMS是否符合MDR的要求,有具体的截止日期嘛? A:无论是已满足器械指令(MDD)要求的器械,还是未满足MDD的器械,按照器械法规(MDR)认证都需要符合MDR的要求。 MDR要求制造商能够展现出有效的质量管理体系。因此,要满足MDR的认证要求,您必须按照法规Article 120的要求,在过渡期内建立合格的质量管理体系。