首页 > 供应商机 > 眼镜架出口MDR2017/745认证 欧盟新版医疗器械法规MDR
眼镜架出口MDR2017/745认证 欧盟新版医疗器械法规MDR
价格:0.00起
上海沙格企业管理咨询有限公司
联系人:袁玲
电话:17701729945
地址:上海市浦东新区陆家嘴街道世纪大道1500号
产品规格:
产品数量:
包装说明:
关 键 词:眼镜架出口MDR2017/745认证
行 业:咨询 管理咨询
发布时间:2023-08-20
级医用防护服、隔离衣、手术衣,欧盟授权代表,签署《欧代协议》;
关于MDR涵盖产品范围和分类规则:器械的分类继续以前的类别,根据风险等级分为四类:I,II a,II b,III。但是,分类规则从18增加到22.具体分类规则的条件和变化如下:关于经济运营商的义务:该法规在章*2条的定义中提出了“经济运营者”的概念。

说到这里 企业应该如何面对新法规的升级呢?按照MDR法规要求。关键的内容包括如下几个方面:
企业的质量管理体系 EN ISO13485:2016 ,产品的型式试验 TYPE TESTING , 产品的技术文件 TECHNICAL CONSTRUCTION FILES要满足这些要求,通常需要咨询机构和咨询师的协助。本公司拥有一支经验丰富的咨询师队伍可以为贵司提供这些服务,包括: 协助贵司建立/升级器械质量管理体系,将MDR法规的内容整合进去 协助贵司确定产品的欧盟协调标准,确认检测实验室的,样品准备以及检测不合格整改的研讨 按照MDR要求协助贵司准备技术文件,包括风险分析报告,评估资料,基本要求检查表等 协助贵司按照欧盟的要求修订说明书和标签,使其满足出口的要求。

2019 年4 月5 日,欧洲议会和理事会正式签发了欧盟关于器械*2019/745 号法规(MDR,EU2019/745),5月5日,欧盟(Official Journal of the EuropeanUnion) 正式发布该法规。2019 年5 月25 日,MDR 正式生效, 替代了原器械指令(MDD,93/42/EEC)和主动植入式器械指令(AIMD,90/385/EEC)。
MDR 由指令升级为法规,提高了对欧盟成员国的约束力,具有直接约束性,*各国转化为本国的法律法规的形式即可落实实施。内容上,MDR 在整合原指令的基础上,大幅提升了有关器械认证的规范和限制,例如关于产品分类规则、器械的可追溯性、性能研究的规范、增加上市后的产品性和有效性的等方面。MDR 共10 章123 条,并附有17 个附录。
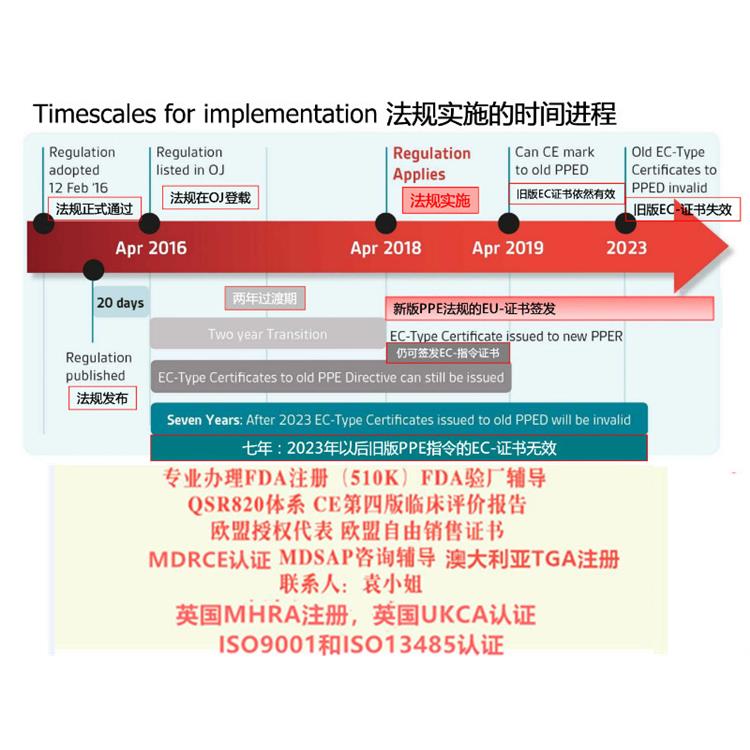
一、关于法规过渡期
MDR 过渡期为3 年,共涉及四个时间点(见表1):
申请欧盟CE认证(CE整套技术文件编订、 CE*四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书
欧盟器械新法规MDR主要变化情况介绍
仅具有根据90/385/EEC 和93/42/EEC 指令签发的证书的器械可投放市场的前提是自MDR 适用之日起,其在设计和预期目的上无显著变化并符合新法规有关市场后监察、市场监察、警戒、经济运营商及器械注册的规定。
通过豁免指令的形式上市,且符合新法规的器械可在2020 年5 月26 日之前投放市场。并可在2020 年5 月26 日前并通知符合新法规的符合性评估机构。公告机构可在2020 年5 月26 日前, 采用合规的符合性评估流程并按照新法规签发证书。

我司注意到新法规主要在以下几点上发生了变化:
1.器械的定义;
2.器械的分类;
3.基本和性能要求;
4.技术文件要求;
5.评价;
6.上市后;
7.Eudamed数据库;
8.对NB公告机构的要求(新法规生效后NB将按照新的资质要求重新进行授权);
9.对高风险器械的新增了要求;
总的说来新的MDR和IVDR加强了体系管理,对高风险设备增加了相关规定比如对于非用途但具有与器械相似特性的设备也将受到新法规的管辖(如用于美容的彩色眼镜),提升了产品对患者的透明度和可追溯性并设立电子数据库(Eudamed)、设备将有一个的识别号这加强其在整个供应链的可追溯性。
SUNGO提醒我们的客户在申请产品CE认证时,在过渡阶段请谨慎考虑是选用新法规还是采用老的指令方案,同时也需要对NB机构的发证进行了解和确认以保证产品在欧盟市场销售的可延续性
我们的核心资源包括分布在**主要经济体的运营网络,具有美国IAS认可资质的实验室,具有ANAB认可资质的认证机构,以及分布**的资源。