手动腔镜吻合器CE*四版临床评价报告 怎么申请
价格:0.00起
产品规格:
产品数量:
包装说明:
关 键 词:手动腔镜吻合器CE*四版临床评价报告
行 业:商务服务 认证服务
发布时间:2023-08-17
法规条款增加,认证评审更加严格,企业的相关信息都会被收集到欧洲器械数据库(EUDAMED);
上市后(PMS) (MDR*83~86条) PMS需收集、记录并分析器械在其整个生命周期内的质量、性能和相关数据,以得出必要的结论,并确定、实施和监测任何预防及纠正措施。
对于公开文献的数据搜集,国内的生产商还有一个常见的错误,即没有预先建立一个搜索的策略,该策略必须囊括所有适用的数据(无论是正面的还是的),而不是选取仅仅是“好的”文献。在*四版的要求中,仅仅获得数据是没有用的,还需要“运用”这些数据来申请产品的性和有效性。“运用”包括:
CE*四版(MEDDEV2.7.1Rev4)报告
评价要进行的是现行有效技术(StateoftheArt)的评估
评价分析器械/等同器械的数据,包括:
的要求(ER1);
风险收益比的要求(ER3);
性能的要求(ER3);
的可接受度(ER6);
以及决定是否需要进行上市后的跟踪;
CE*四版(MEDDEV2.7.1Rev4)报告
在分析的过程中,*四版更注重引入“统计分析”的方法,包括:
数据评估和加权(*9节和附录6)
数据分析和证实符合性(*10节和附录7)
*四版的原则还对“”提出了明确的要求,包括资质要求以及“利益声明”的要求。资质要求每一个评审人员具有高等学历以及5年的工作经历(或不具备高等学历时,10年工作经历)。工作经历并不单纯的指的经历。常见的模式是评价由生产商的产品准备,因为没有人比生产商自己更了解他们的产品,那么对于产品来说,其工作经历应当是与改产品研发相关的经历。此外,评价报告应当由进行审核,那么对于来说,需要有相关的工作经历,而且其工作经历应该确保其熟悉该产品的适用。
CE*四版(MEDDEV2.7.1Rev4)报告
按照*四版原则的要求,评价报告应当包含以下内容:
(一)概要
(二)评价的范围
(三)评价的背景:当前的知识,新的技术水平
(四)评价的设备
4.1评价的类型
4.2等同性说明
4.3制造商产生和持有的数据
4.4来自于文件的数据
4.5数据的总结和审核
4.6数据的分析
(五)结论
(六)下次评价的日期
(七)日期和签名
(八)负责评价的评价者的资质
(九)参考
评价报告还应当确定并论证定期更新的频率:
如果有严重风险或者没有良好的建立风险,则至少每年1次
如果没有严重风险并且已经良好地建立了风险,则2-5年一次
当上市后数据显示对现有的评价有影响时需要主动更新!
CE*四版(MEDDEV2.7.1Rev4)报告
对于生产商的一些建议
1.相比于*三版,*四版给予了更多的指南,生产商应从中考虑问题力求评估文件完整性和科学性。
2.生产商应关注*四版相比与*三版的差,使评估文件符合新法规的要求。
3.生产商除了关注CE相关法规外,也应关注本国和其他拟出口的评估要求。随着食品品监督和CE以及FDA法规当局的沟通利益密切,可以看出监局的评估指南,对于CE的评估指南有一定的参考和借鉴意义。
4.生产商应将评估程序纳入质量体系,并应综合考虑拟销售的相关试验/评估的法规要求。
关于MEDDEV2.7.1Rev4,SUNGO可以协助您:
1、协助建立评价程序;
2、建立评价方案
3、寻找等同产品,进行等同分析;
4、搜索文献及其他数据;
5、数据分析;
6、完成评价报告。

对于需要公告机构介入的器械,符合性声明的签署通常是符合性评定程序的一步。在未取得公告机构签发的CE证书之前,制造商无法签署正式的DOC文件。不过制造商可以先起草一份DOC的草案提供给公告机构审查。而对于*公告机构介入的器械,制造商在法规所要求的产品符合基本要求的证据准备充分后,即可签署DOC。 DOC作为法规要求的重要文件,制造商应该按照质量管理体系中文件控制程序的要求进行管控。DOC中任何内容发生了变更,则需重新签发。特别需要注意的是,对于由公告机构发证的产品,DOC中任何内容的变更,都需得到原发证公告机构的评审和批准。

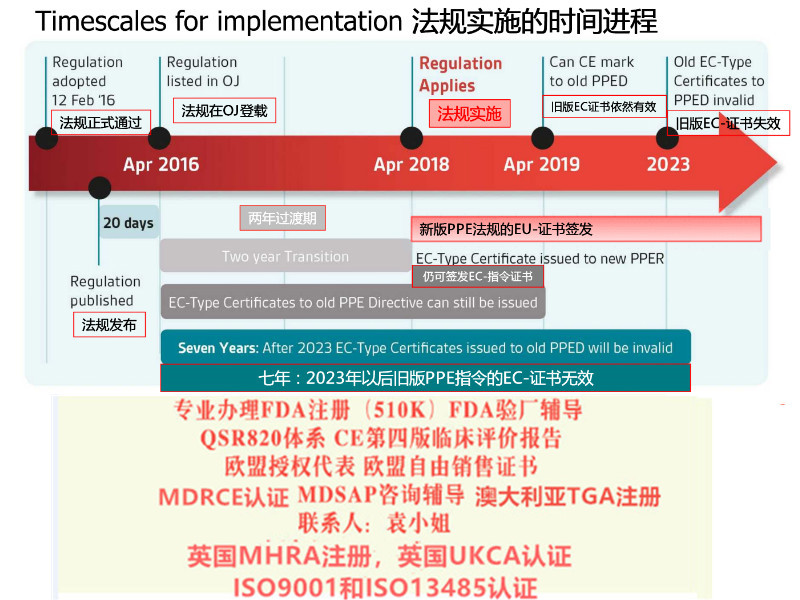
2017年5月5日欧盟发布OfficialJournal在这里我司需要特别说明的是欧盟此次是直接发布的Regulation(法规)而相比较之前的Directive(指令)其区别在于:提高了文件的约束力,发布立即在欧盟成员国生效并成为有约束力的法律,此次的Regulation*向Directive那样需要经过成员国转化成当地法律法规去落实实施。器械法规(MDR)转换期为3年,2020年5月4日起强制实行。
ISO13485认证,CE评估报告编写,FDAQSR820等产品出口的相关认证


